Publication
Article
Special Issues
Hemophagocytic Lymphohistiocytosis in a Patient With Nodular Lymphocyte–Predominant Hodgkin Lymphoma: A Case Report
Author(s):
This is the first case report of hemophagocytic lymphohistiocytosis complicating a nodular lymphocyte–predominant Hodgkin lymphoma in an adult treated with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone.
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a rare and rapidly progressing clinical syndrome with unknown incidence in adults that results from severe immune activation. Characteristic findings include fever, hepatosplenomegaly, lymphadenopathy, pancytopenia, rash, and neurological symptoms with elevated triglycerides, ferritin, and hypofibrinogenemia.
HLH is often described as primary hemophagocytic lymphohistiocytosis (inherited), with underlying genetic abnormality, or secondary (acquired) to another underlying condition such as a viral infection, an autoimmune disease, or a malignancy.
To the best of our knowledge, this is the first case report of HLH complicating a nodular lymphocyte—predominant Hodgkin lymphoma (NLPHL) in an adult treated with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone. The NLPHL showed the additional, unusual feature of cyclin D1 expression, which, to our knowledge, also has not been previously reported.The patient died after treatment failure following 3 regimens of therapy.
High clinical suspicion of HLH should prompt immediate immunochemotherapy to reverse the otherwise high likelihood of a fatal outcome.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a syndrome caused by an exaggerated inflammatory response and cytokine defect resulting in organ infiltration by activated cytotoxic T lymphocytes and histiocytes.1,2 Disproportionate levels of TNF-alpha; interleukins (IL) 4, 6, 8, 10, and 12; and soluble IL-2 receptor (sCD25) have been recorded in patients with HLH in combination with impaired/absent natural killer (NK) cell activity.3 The aforementioned parameters can aid in distinguishing this syndrome from etiologies with similar manifestations. Primary HLH, which generally affects infants and young children, is attributed to defects in perforin function. The incidence of secondary HLH in adults is unknown. Secondary HLH, either as a presenting phenomenon or developing during the course of a primary disease, has a broad range of associations, including infections (eg, Epstein-Barr virus [EBV] and cytomegalovirus [CMV]), rheumatic diseases, and malignancies, such as lymphoma).
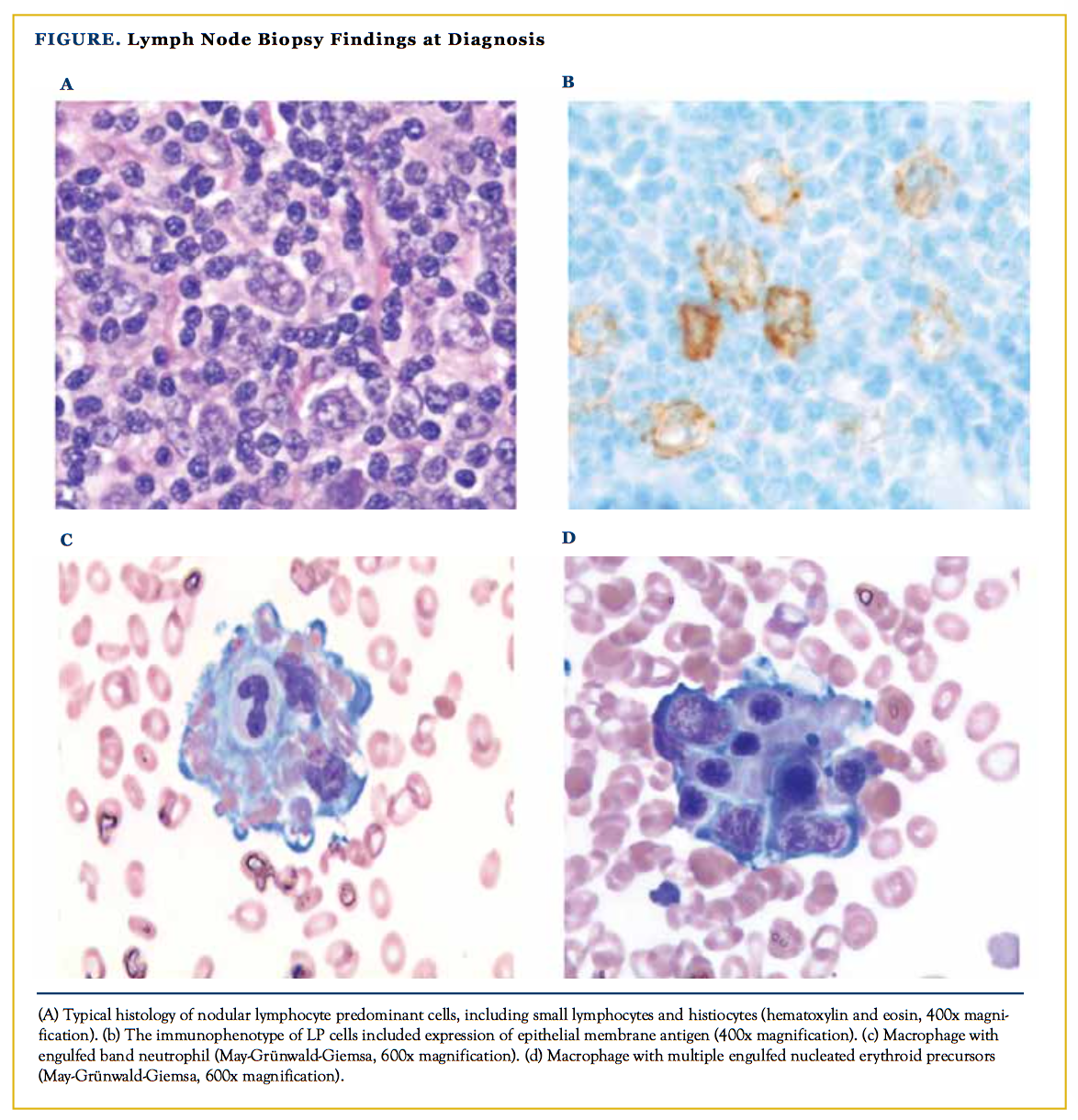
Clinical and laboratory findings are characteristic yet nonspecific, which can delay the diagnosis. Findings include prolonged fever, hepatosplenomegaly, lymphadenopathy, pancytopenia, rash, and neurological symptoms, usually with a rapidly progressing course.1,4 Classic laboratory features include elevated triglycerides, markedly elevated ferritin levels, hypofibrinogenemia, and abnormal liver function tests. Bone marrow examination often reveals characteristic hemophagocytes— histocytes with engulfed hematopoietic elements, including nucleated erythroid precursors, white blood cell precursors, and platelets—which are best depicted in marrow aspirate smears.
HLH is a challenging diagnosis due to its rarity and it’s nonspecific presentation, and laboratory findings. Prompt recognition and diagnosis is important, given the necessity of early treatment to assure the best possible outcome. The main goal of therapy, especially when HLH is associated with malignancy, is to direct therapy toward the underlying condition while attempting to control the amplified inflammatory reaction.4
Case Presentation
Our patient is a 26-year-old Caucasian man with past medical history significant for nodular lymphocyte—predominant Hodgkin lympoma (NLPHL) stage IIIBs (lymph node involvement above and below the diaphragm with splenic involvement). Lymph node biopsy findings at diagnosis were typical of NLPHL, including macronodular architecture with a predomi- nance of small lymphocytes, lesser numbers of histocytes, and a minor population of admixed lymphocyte–predominant (LP) cells (Figure). LP cells expressed CD45, CD20, CD79a, BCL6, and epithelial membrane antigen, and lacked expression of CD10, CD15, and CD30. Interestingly, cyclin D1 was also expressed by cells consistent with LP cells (large, pleomorphic cells having highly lobated nuclei). The patient completed 6 cycles of rituximab, doxorubicin, vincristine, and predni- sone (R-CHOP) with resolution of the presenting symptoms following 2 cycles. The patient achieved unconfirmed complete remission at the end of therapy due to a small residual node on the left infraclavicular region.
After 4 months, a positron emission tomography/computed tomography (PET/CT) scan to follow up on the residual findings described above showed persistent local uptake. This was removed by surgery, with pathology showing extensive necrosis. A second PET/CT scan showed no further abnormal findings. One year after completion of therapy, the patient was seen in the office with recurrent symptoms (mainly night sweats) and was found to have pancytopenia upon blood count. A third PET/CT scan showed an enlarged left supraclavicular mass (4.3 × 1.3 cm) with a standardized uptake value of 13. Laboratory investigation showed the following results: white blood cell count, 1.2 × 103/mcL; absolute neutrophils, 0.6 × 103/mcL; hemoglobin, 11 g/dL; mean corpuscular volume, 87 fL; platelet count, 130.000/mcL; ferritin, 1387 ng/mL; alanine transaminase, 122 U/L; aspartate transaminase, 121 U/L; total bilirubin, 2.5 mg/dL; alkaline phosphatase, 303 U/L; albumin, 3.5 g/dL; prothrombin time, 18 seconds; creatinine, 0.8 mg/ dL. Bone marrow biopsy showed normal trilineage hematopoie- sis and increased cellularity (70%) without evidence of lympho- matous involvement. The patient underwent local radiation therapy to the supraclavicular area.
Six weeks later the patient was hospitalized for possible sepsis, because he presented with fever, splenomegaly, and cytopenias. The laboratory investigations showed hypertriglycerdemia and hyperferritenemia, with elevated liver enzymes. A bone marrow biopsy showed scattered histocytes displaying hemophagocytosis of both myeloid and erythroid precursors, and normal to increased cellularity with no evidence of lymphoma. He was treated with oral prednisone 100 mg daily and rituximab 375 mg/m2 intravenous once weekly for 4 weeks with mild improvement in his symptoms. He required intravenous hydration on 2 occasions following drenching sweating.
The patient developed severe jaundice due to progressive hepatic failure, acute kidney injury, and bloody diarrhea after a few weeks, which required readmission. Salvage therapy with alemtuzumab, per standard protocol, and dexamethasone 40 mg intravenous daily for 4 days was initiated with no significant response. The patient was subsequently treated with pentostatin 4 mg/m2 without a response. His condition further deteriorated with the development of disseminated intravascular coagulation. The patient was transferred to hospice and died shortly thereafter.
Discussion
NLPHL is a rare type of Hodgkin lymphoma (HL), accounting for just 5% of cases. Unique findings include variants of Hod- gkin Reed-Sternberg cells (HRS cells), also known as LP cells. These display an intact B-cell program, expressing pan B-cell markers including CD20 and CD79a; express CD45; and typ- ically lack expression of CD15 and CD30, distinct from HRS cells found in classical HL. Morphologic and immunopheno- typic features in this case were typical of NLPHL with notable cyclin D1 expression by LP cells.
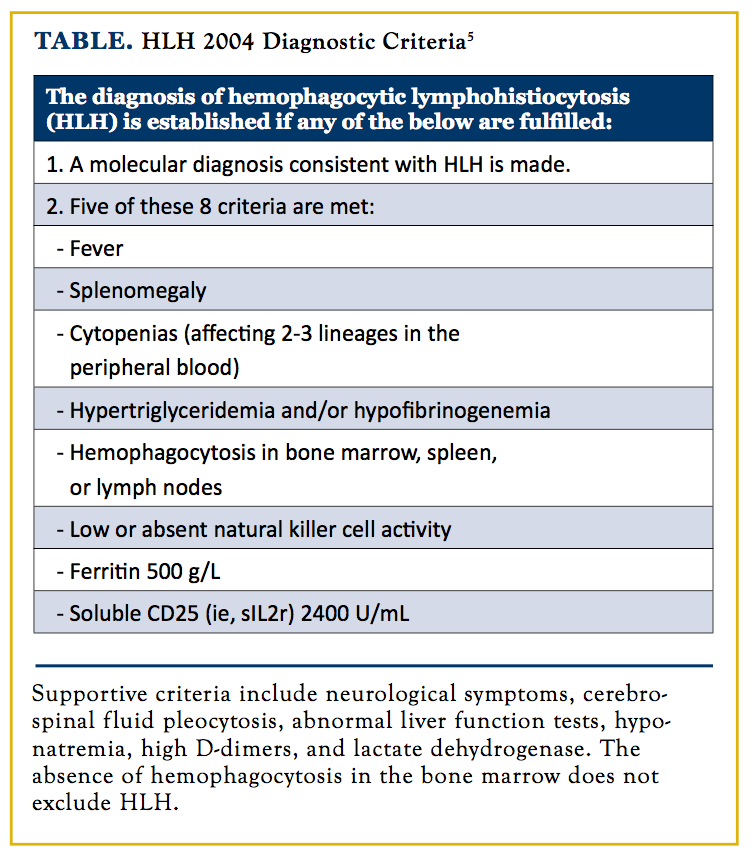
We are unaware of any previous case report of HLH developing in the course of NLPHL. It can be argued that our patient had HLH at initial diagnosis of NLPHL but it was not pursued. The poor prognosis of HLH mandates initiation of therapy as soon as possible, not wait for confirmation investigations when clinically suspected. Updated diagnostic criteria used in the HLH-2004 trial by the Histiocyte Society are described in the Table.5
In our case, diagnosis of HLH was delayed at least 6 weeks following development of bicytopenia, fever, splenomegaly, and a markedly elevated ferritin. Four of 5 criteria required for HLH diagnosis were met. NK activity, soluble CD25, triglycerides, and fibrinogen were not assessed at that time, which might have been useful investigations. The patient was actually fulfilling the proposed diagnostic criteria, as HLH-2004 criteria are not specific and mainly used for clinical trials.5 The proposed criteria are 3 of 4 clinical manifestations (fever, splenomegaly, cytopenias, and hepatitis) in addition to 1 of 4 investigational findings (hemophagocytosis, high ferritin, low fibrinogen, and absent/decreased NK cell function).
In a series of 34 patients with HL—associated hemophagocytic syndrome, most patients with were male, predominantly lymphocyte depleted, and mixed cellularity subtypes of classical HL (85%) with EBV involvement (95%).6 None of these patients had NLPHL (14 patients with unknown type). Treatment of triggering infection improves outcomes. Described culprits include EBV, CMV, HIV, herpes simplex virus, varicella zoster virus, and Leishmania. Our patient was EBV immunoglobulin (Ig) G positive and IgM negative. In adult patients, NK/T-cell degranulation, activity studies, and genetic testing can be used to explore the possibility of late onset of a congenital variety, which was not performed in our case.
Malignancy-associated HLH has a worse prognosis compared with other secondary forms and there are no evidence-based guidelines to direct therapy.7 A common practice is to initiate treatment aimed at the presenting malignancy and to add other measures to control the hyperimmune reaction based on the response to initial therapy.4
The Histiocyte Society initiated a prospective international collaborative therapeutic study in 1994 (HLH-94) that aimed to improve survival. Standard treatment for primary HLH (HLH- 94 protocol) includes 8 weeks of initial therapy, consisting of etoposide (150 mg/m2 twice weekly for 2 weeks and then weekly) and dexamethasone (initially 10 mg/m2 for 2 weeks followed by 5 mg/m2 for 2 weeks, 2.5 mg/m2 for 2 weeks, 1.25 mg/m2 for 1 week, and 1 week of tapering). The initial therapy was followed by a continuation therapy and bone marrow transplant for patients with persistent disease or known familial disease.
Patients with unresolved nonfamilial disease stopped therapy. The continuation therapy included dexamethasone and etoposide alternating every second week in combination with daily oral cyclosporin. Intrathecal methotrexate was administered for patients with progressive neurological symptoms or consistent abnormal cerebrospinal fluid findings. The HLH-94 study enrolled 119 patients who were younger than 15 years of age. The study excluded 6 patients who were subsequently found to have specific underlying disorders (4 with hematolymphoid malignancies). This study is the only known study to date to show an improved survival with an estimated 3-year probability of overall survival of 55% (95% CI, 46%-64%). However, given the patient demographics, we speculate that the majority of the patients enrolled had a familial predisposition.8
Rituximab showed good response if the trigger was EBV infection.8,9 So et al also reported a case of HLH and autoimmune hemolytic anemia associated with systemic lupus erythematosus successfully treated with rituximab that was refractory or intolerable to conventional therapy.10 During the second presentation, our patient did not respond to 4 weeks of rituximab and steroid treatment. He was also treated with cyclosporine, dexamethasone, and etoposide without significant improvement.
Alemtuzumab may be appropriate, instead of etoposide, in the case of liver failure, clinical deterioration, or no improvement within 2 to 3 weeks of HLH-94 therapy.11-13 Cyclophosphamide, doxorubicin, vincristin, prednisone (CHOP) therapy for 8 weeks in 17 adult patients showed a 2-year overall survival of 44%14; however, our patient received R-CHOP as an initial therapy for NLPHL and repeat of the same regimen was not entertained. Unfortunately, most trials in the literature included limited numbers of patients and the various regimens used in those trials were not compared with HLH-94-based therapy, which remains the standard of care. Although hematopoietic stem cell transplantation in familial HLH has been shown to improve outcomes, its role in secondary types has not been defined.3
Conclusion
Despite improvements in the diagnosis and treatment of HLH, this entity remains an often-fatal clinical syndrome that requires prompt recognition and intervention. HLH can be associated with NLPHL, and clinician awareness of early manifestations is crucial to avert fatal outcomes. More effective therapies are also required to improve patient outcomes and should be a focus of further studies.
Acknowledgements: None.
Financial disclosures: None.
Author affiliations: Delshad Ahmad, Mohamed Akkad, and Anas Al-Janadi are with Michigan State University; Brian Olsen is with William Osler Health System.
Address correspondence to: Anas Al-Janadi, MD, Breslin Cancer Center, Michigan State University, 401 W. Greenlawn Ave, Lansing, MI 48910.
References
- Okamoto M, Yamaguchi H, Isobe Y, et al. Analysis of triglyceride value in the diagnosis and treatment response of secondary hemophagocytic syndrome. Intern Med. 2009;48(10):775-781.
- Fukaya S, Yasuda S, Hashimoto T, et al. Clinical features of haemophagocytic syndrome in patients with systemic auto- immune diseases: analysis of 30 cases. Rheumatology (Oxford). 2008;47(11):1686-1691. doi: 10.1093/rheumatology/ken342.
- Mehta RS, Smith RE. Hemophagocytic lymphohistiocytosis (HLH): a review of literature. Med Oncol. 2013;30(4):740. doi: 10.1007/s12032-013-0740-3.
- Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118(15):4041-4052. doi: 10.1182/ blood-2011-03-278127.
- Alexandra H. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology Am Soc Hematol Educ Program. 2009;127-131. doi: 10.1182/asheducation-2009.1.127.
- Filipovich AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology Am Soc Hematol Educ Pro- gram. 2009;127-131. doi: 10.1182/asheducation-2009(1):127.
- Ménard F, Besson C, Rincé P, et al. Hodgkin lymphoma associated hemophagocytic syndrome: a disorder strongly correlated with Epstein-Barr virus. Clinical Infect Dis. 2008; 47(4):531-534. doi: 10.1086/590152.
- Machaczka M. Hemophagocytic lymphohistiocytosis in adults. Ups J Med Sci. 2013;118(3): 201-203. doi: 10.3109/03009734.2013.795634.
- Henter JI, Samuelsson-Horne A, Aricò M, et al; Histocyte Society. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow trans- plantation. Blood. 2002;100(7):2367-2373.
- Al Asad O, Salam A, Mannem S, Ninan M, Markowitz A, Jana B. Alternative therapy for Epstein-Barr virus related hemophagocytic lymphohistiocytosis. Case Rep Oncol Med. 2015;2015:508387. doi: 10.1155/2015/508387.
- So MW, Koo BS, Kim YJ, Kim YG, Lee CK, Yoo B. Successful rituximab treatment of refractory hemophagocytic lymphohistiocytosis and autoimmune hemolytic anemia associated with systemic lupus erythematosus. Mod Rheumatol. 2014;24(5):855-857. doi: 10.3109/14397595.2013.874740.
- Jiang L, Yuan CM, Hubacheck J, et al. Variable CD52 expression in mature T cell and NK cell malignancies: implications for alemtuzumab therapy. Br J Haematol. 2009;145(2):173-179. doi: 10.1111/j.1365-2141.2009.07606.x.
- Strout MP, Seropian S, Berliner N. Alemtuzumab as a bridge to allogeneic SCT in atypical hemophagocytic lymphohistiocytosis. Nat Rev Clin Oncol. 2010;7(7):415-420. doi: 10.1038/nrclinonc.2010.40.
- Marsh RA, Allen CE, McClain KL, et al; Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer. 2013;60(1):101-109. doi: 10.1002/pbc.24188.
- Shin HJ, Chung JS, Lee JJ, et al. Treatment outcomes with CHOP chemotherapy in adult patients with hemophagocytic lymphohistiocytosis. J Korean Med Sci. 2008;23(3):439-444. doi: 10.3346/jkms.2008.23.3.439.